
Home > News > Key Risks in Medical Device Development
Key Risks in Medical Device Development
Key Risks in Medical Device Development
Understanding and mitigating these risks during medical device development can reduce your development timeline and ensure success.
By: Travis Schneider
Business Development Manager, Aerotech
Medical innovation is exciting. It's thrilling to think a new therapy, device or product can improve the quality of or save lives. Who can't get behind that? As a result, medical technology applications often rank highest amongst new startup ideas. With an ever-aging global population, there is increasing demand for healthcare solutions, advancements in technology and more efficient, accessible medical devices and services throughout the world.
However, developing a medical device in the modern era can be akin to navigating a minefield. Medical device development has inherent risks that encompass technical, regulatory and clinical factors. Mitigating these risks demands meticulous planning, rigorous testing and vigilant risk management throughout the entire product lifecycle to ensure safety and efficacy for patients and healthcare providers. Given these many critical considerations, medical device development is more complex than ever.
As a result, medical device manufacturers need to thoroughly address many risks associated with developing a new device. This paper will highlight critical factors medical device manufacturers need to consider as development begins and throughout the product's life cycle.
The Role of Classification
Medical product development does not start with design. There are many aspects to consider earlier in the development process, including design for manufacturing, testing, verification and validation. The scrutiny required in examining each of these stages typically depends on the type or class of the medical device being developed.
Medical devices are classified based on the level of regulatory control required to ensure their safety and efficacy. Classification systems may vary slightly among different countries or regions, but the one established by the United States Food and Drug Administration (FDA) is commonly used. While the European Union puts medical devices into one of four classes (Class I, Class IIa, Class IIb and Class III), the FDA has three main classes [1]:
Class I Medical Devices
• Low- to moderate-risk devices
• Simple in design and typically do not pose a significant risk to patients
• Examples include bandages, examination gloves and some handheld surgical instruments
• Generally subject to the least regulatory control
Class II Medical Devices
• Moderate- to high-risk devices
• More complex than Class I devices and may have higher risks associated with their use
• Examples include powered wheelchairs, infusion pumps and certain diagnostic devices
• Subject to more stringent regulatory controls, including special labeling requirements and post-market surveillance
Class III Medical Devices
• High-risk devices
• Typically used to sustain or support life, have a significant risk of illness or injury, or are new and innovative with no existing equivalent devices
• Examples include implantable pacemakers, heart valves and some advanced diagnostic imaging devices
• Subject to the highest level of regulatory control, including pre-market approval
These classifications help regulatory authorities determine the appropriate level of oversight needed during a device's approval process. This ensures medical devices meet specific safety and performance standards before they can be marketed or used in healthcare settings.
Requirements Development and Management
Requirements development and management may be the single most important aspect of developing a new medical device. It is a critical process that involves defining, documenting and controlling a medical device's specifications and characteristics throughout its development lifecycle. The requirements serve as the foundation for designing, manufacturing and validating the device to ensure it meets its intended use and regulatory standards.
Careful attention to requirements development and management is essential for delivering safe, effective, high-quality medical devices. Without rigorously developing, refining and tracking requirements, developers are likely setting a new product up for failure. This process can be time consuming and even painful for a design team, but skipping it would expose them to several regulatory, commercial and technical challenges.
Here are some key elements of the requirements development and management process:
Requirements Discovery
This is the initial stage where stakeholders including end users, clinicians, regulatory experts and engineers gather to identify the medical device's needs and expectations. This involves conducting interviews, workshops and surveys to collect valuable input. It is important to understand the general problem this new device will solve and define the optimum outcomes it will help to achieve for the patient. It's important to avoid biasing prospective end users when conducting these interviews – there is a natural tendency for developers to show a solution rather than uncover a true unmet need.
Requirements Documentation
Once requirements are identified, they need to be systematically documented. Proper documentation ensures clarity, traceability and alignment with regulations. Requirements documents may include user requirements specifications, design input requirements and functional specifications. Requirements documentation should also involve a number of roles in order to ensure the right checks and balances are in place. Product or marketing managers often document requirements in a less technical fashion. Engineering teams then translate those requirements into more detailed or technical requirements that development teams can work toward. Marketing and engineering teams must communicate to ensure this translation accurately captures user needs in the proper context.
Validation and Verification
Requirements must be verified and user needs must be validated to ensure they are accurate, complete and aligned with the device's intended use. Validation ensures the device meets its intended purpose, while verification checks that the device design complies with specified requirements.
Traceability Management
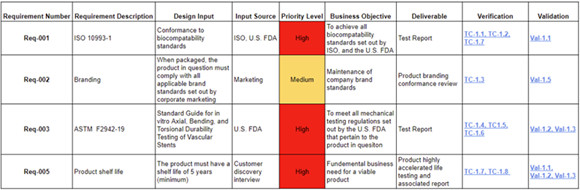
Traceability is the ability to link requirements to design, verification and validation activities. A robust traceability matrix like the one shown in Table 1 helps to ensure each requirement is addressed during the development process and provides a basis for regulatory submissions. Maintaining this traceability matrix throughout the development process will save significant time and effort during later phases of releasing the product to market.
Table 1: An example of a requirements traceability matrix.
Change Control
Throughout the device development lifecycle, requirements may change as a result of new insights, feedback or regulatory updates. Change control processes ensure that all changes are managed, evaluated for impact and properly documented to maintain the integrity of the device's development.
Risk Management
Identifying and managing the medical device's risks and potential harms is essential for mitigating associated hazards and ensuring patient safety. A risk register is an essential tool that helps design teams manage risk throughout the development process, and it provides a more analytical approach to addressing the highest risk element early in the development process. This template [2] will be useful fordevelopers who don’t have a standard risk register or want to improve their current approach.
Regulatory Compliance
Medical device requirements must align with relevant regulations, standards and guidelines like those set by the FDA, ISO 13485 and the European Medical Device Regulation. Demonstrating compliance with these regulations is crucial for obtaining regulatory approvals.
User Feedback Loop
Establishing a feedback loop with stakeholders, end users and other relevant parties helps in refining and updating the requirements based on real-world experiences and evolving needs. This is critical for the product’s technical refinement and commercial validation.
Requirements Verification Testing
As part of the design verification process, medical device manufacturers must perform tests to demonstrate the device meets its specified requirements. Verification testing confirms the device functions as intended and complies with applicable standards.
Design Controls
Properly managing requirements is an integral part of the overall design controls process, which ensures a systematic approach to developing medical devices that meet user needs and regulatory requirements.
Effective requirements development and management play a crucial role in successfully bringing medical devices to market, fostering patient safety and enhancing overall product quality and performance. It is an iterative process that continues throughout the device’s lifecycle. Poor requirements management will result in significant project delays and increase the likelihood of significant risks to the patient – as well as potential for recall and litigation. Often, a strong project management office will help to champion and steward requirements management by providing structure, oversight and effective communication throughout the development process.
These aspects are particularly important to manufacturers who choose to either outsource or partner with third parties to develop a device. It’s often critical that a partner organization has an appreciation for rigorous requirements development and understands how those requirements are interconnected. Ideal partner organizations will mirror thecapabilities of the medical device's original equipment manufacturer, have a detailed understanding of the industry, staff a dedicated project management team and maintain their own quality management system. They should understand the importance of requirements and be able to properly translate them to the necessary scope.
Regulatory Risk
Regulatory compliance risk in medical device development refers to the challenges and uncertainties related to obtaining the appropriate regulatory approvals and compliance with the applicable laws, regulations and standards set forth by the governing body in a market (e.g. FDA in the United States). Bringing a medical device to market involves navigating a complex and ever-evolving landscape of regulatory requirements, and failure to address these risks adequately can result in delays, increased costs or even the inability to market the device.
One of the critical risks is classification risk. Determining the correct regulatory class for the device is essential, as misclassifying the device could lead to incorrect submission pathways or insufficient data support.
Furthermore, the regulatory landscape is constantly changing, so medical device companies must keep abreast of frequent updates and changes in regulations to ensure compliance throughout the development process. Seasoned regulatory professionals have grown accustomed to this climate and often plan with contingencies in mind.
In addition to a strong regulatory department, medical device manufacturers should also staff a strong quality department to help navigate compliance by maintaining a robust quality management system (QMS). Neglecting to implement an effective QMS may lead to non-compliance findings during regulatory audits. Often these two departments work closely to produce a design history file (DHF), a comprehensive and organized compilation of documentation that captures the device's design and development history. The DHF is a critical regulatory requirement for medical device manufacturers and serves as a permanent record of the product's design control activities and decision-making processes throughout its development lifecycle.
Proper submission is also key – obtaining regulatory approvals can be a lengthy process, potentially impacting market entry and straining financial resources if delayed. Insufficient preclinical or clinical data to support safety and efficacy claims can result in rejected regulatory submissions or requests for additional studies, further delaying approvals and resulting in project cost overruns. Moreover, global variability in regulatory frameworks demands strategic planning to navigate different countries' requirements for international market access.
Even after the device is on the market, medical device manufacturers must establish effective post-market surveillance systems to monitor and report adverse events and malfunctions. Failure to do so may lead to regulatory non-compliance and potential product recalls. Proper labeling and marketing are also essential for compliance. Misleading claims or inadequate instructions can result in regulatory penalties and harm a company's reputation. Other post-release issues can also stem from neglecting human factors and usability considerations, and these can result in use errors and additional regulatory findings.
Cost Risk
Cost risk is the potential for unforeseen expenses that can increase the overall cost of production. Managing cost risk is essential for medical device manufacturers to ensure profitability, financial stability and market competitiveness.
Several key factors contribute to cost risk in medical device manufacturing. The first is material costs, including raw materials and components, which can fluctuate due to changes in market conditions, supply chain disruptions or geopolitical factors. Dependency on specific suppliers introduces supply chain risks. Disruptions, delays or quality issues with suppliers can lead to increased costs or production delays. Sudden price increases or shortages of critical materials can significantly impact manufacturing costs. In recent years, manufacturers have also had to contend with the impact of economic changes like inflation or recessionary periods on manufacturing costs.
Labor costs also play a crucial role in manufacturing expenses. Wages, benefits and training expenses can vary depending on location and market conditions. Changes in labor costs, workforce availability or labor regulations can affect overall manufacturing expenses. Automation and robotics can often be used to augment or manage labor costs, but often some degree of manual intervention is required for Class III products.
Finally, regulatory compliance itself is a significant cost driver. Medical device manufacturers must invest in research, testing, documentation and certification to meet necessary standards, adding to overall manufacturing expenses. Similarly, quality control and testing are another important aspect of cost risk. Maintaining a robust quality control process and investing in sophisticated testing equipment and skilled personnel to ensure the device's safety and efficacy can increase manufacturing costs.
To manage cost risk effectively, medical device manufacturers must conduct comprehensive cost analyses, develop contingency plans and maintain flexibility in their supply chain and manufacturing processes. Adopting lean manufacturing practices and continuous improvement initiatives can help to optimize costs while ensuring high product quality and compliance with regulatory requirements. Additionally, close collaboration with suppliers and a proactive approach to identifying and addressing cost drivers can mitigate cost risk and enhance the overall financial health of the medical device manufacturing business.
Schedule Risk
Given the competitive landscape of the medical technology industry, speed to market is key. Delays or disruptions in the product development timeline can affect the planned schedule for bringing the device to market, corresponding revenue projections and the business case analysis. Managing schedule risk is essential if medical device manufacturers are going to meet regulatory deadlines, market demands and financial goals.
Here, too, it is ideal to have a strong project management team to handle the complex schedule for bringing a product to market. As shown in Figure 1, an example schedule is often riddled with interdependencies and gating factors that cannot be expedited (e.g. regulatory submissions, biocompatibility testing, accelerated life testing, etc.). Without careful oversight and planning, the project team could experience delays or periods of limited resource utilization while certain pacing items occur.
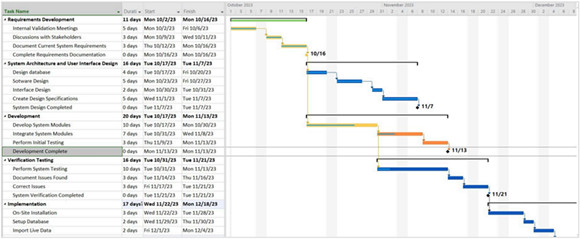
Figure 1: An example of a Gantt chart.
Many items can impact the schedule. Examples mentioned above include regulatory submissions, supplier delays, testing and validation. Changes in technical scope and requirements late in the development process can also throw a significant wrench in plans as they may render many of the completed tasks invalid.
To manage schedule risk effectively in medical device manufacturing, companies should use proactive project management practices like the following:
- Develop a detailed project plan with clear milestones and deadlines.
- Identify potential risks early in the development process and develop contingency plans, especially for highest risk elements.
- Regularly monitor progress and adapt the schedule as needed to address any delays or issues that arise.
- Maintain open communication among all stakeholders to ensure alignment and quick resolution of any roadblocks.
- Seek expert advice and leverage experience from past projects to anticipate and mitigate potential schedule risks.
By taking a proactive and adaptive approach to schedule risk management, medical device manufacturers can increase the likelihood of meeting project timelines and successfully bringing safe and effective products to market on schedule.
About the Author

Travis Schneider is Aerotech's business development manager for advanced manufacturing market segments, including electronics manufacturing, laser processing, medical technology, data storage and precision manufacturing. He has 13+ years of experience in precision automation and robotics, holding roles in applications engineering, field sales, product management and business development. He has worked alongside several leading medical instrument builders, medical device OEMs, and surgical robotic companies to develop and release products to the market. Travis earned his bachelor's degree in Mechanical Engineering from the Milwaukee School of Engineering. His expertise and passion for innovation make him an invaluable resource for partners seeking to push boundaries in precision automation for advanced manufacturing.
References:
[1]“Classify Your Medical Device,” U.S. Food & Drug Administration, February 7, 2020,
https://www.fda.gov/medical-devices/overview-device-regulation/classify-your-medical-device.
[2]“Risk Register Template,” Leading Agile, 2023,
https://www.leadingagile.com/wp-content/uploads/2015/05/Risk-Register-Sample.xlsx.
Contact us
Aerotech, Inc.
Tel: +86 (21) 5508 6731
E-mail: sales@aerotech.com
To manage schedule risk effectively in medical device manufacturing, companies should use proactive project management practices like the following:
- Develop a detailed project plan with clear milestones and deadlines.
- Identify potential risks early in the development process and develop contingency plans, especially for highest risk elements.
- Regularly monitor progress and adapt the schedule as needed to address any delays or issues that arise.
- Maintain open communication among all stakeholders to ensure alignment and quick resolution of any roadblocks.
- Seek expert advice and leverage experience from past projects to anticipate and mitigate potential schedule risks.
By taking a proactive and adaptive approach to schedule risk management, medical device manufacturers can increase the likelihood of meeting project timelines and successfully bringing safe and effective products to market on schedule.
About the Author
Travis Schneider is Aerotech's business development manager for advanced manufacturing market segments, including electronics manufacturing, laser processing, medical technology, data storage and precision manufacturing. He has 13+ years of experience in precision automation and robotics, holding roles in applications engineering, field sales, product management and business development. He has worked alongside several leading medical instrument builders, medical device OEMs, and surgical robotic companies to develop and release products to the market. Travis earned his bachelor's degree in Mechanical Engineering from the Milwaukee School of Engineering. His expertise and passion for innovation make him an invaluable resource for partners seeking to push boundaries in precision automation for advanced manufacturing.
References:
[1]“Classify Your Medical Device,” U.S. Food & Drug Administration, February 7, 2020,
https://www.fda.gov/medical-devices/overview-device-regulation/classify-your-medical-device.
[2]“Risk Register Template,” Leading Agile, 2023,
https://www.leadingagile.com/wp-content/uploads/2015/05/Risk-Register-Sample.xlsx.
Contact us
Aerotech, Inc.
Tel: +86 (21) 5508 6731
E-mail: sales@aerotech.com
Related News
- Lilac Biosciences and Soin Neuroscience Announce Publication of Clinical Review 7/24/2026
- Matexcel Launches Mid-Year Special Promotion to Support Materials Science Resear 7/24/2026
- Epidermal Growth Factor (EGF): A Revolutionary Molecule from Basic Biology to Re 7/24/2026
- How DPP-4 Research Becomes the National Natural Science Foundation’s "Golden Tra 7/23/2026
- Potent Payloads in ADCs: Enhancing Targeted Cancer Therapy through Payload Techn 7/22/2026
- Axiom Biosciences Announces Intention to Become the First U.S. Biotech to List o 7/22/2026
- Win an INTEGRA Biosciences PIPETBOY GENIUS Pipet Controller! 7/21/2026
- PTEN Companion Diagnostic Antibodies Direct Precision Cancer Therapy 7/21/2026
- Illumina Expands Billion Cell Atlas Program with New AI Drug Developers 7/20/2026
- PTEN Antibody Decodes Tumor Suppressor Inactivation and Precision Therapy 7/20/2026